| pA2 online © Copyright 2004 The British Pharmacological Society |
154P
University of Newcastle Winter Meeting December 2004 |
The impact of genetic polymorphisms of drug metabolising enzymes on pharmacological response: an integrated model G. L. Dickinson, N. J. Proctor, M. S. Lennard, G. T. Tucker & A. Rostami-Hodjegan. Academic Unit of Clinical Pharmacology, The University of Sheffield, The Royal Hallamshire Hospital, Sheffield, |
|
Functional allelic variants of drug metabolizing enzymes (e.g. cytochromes P450; CYPs) contribute to inter-individual variation in plasma drug concentrations. However, the consequence of these genetic variations for pharmacological response is unclear [1,2]. Prospective clinical trials demonstrating the effects of CYP genotype on pharmacological or toxicological response to drugs are virtually absent from the literature, and findings from retrospective studies and case reports are not always consistent. This may be due to difficulty in estimating the power of such studies a priori, as the latter requires combination of estimates of pharmacokinetic (PK) and pharmacodynamic (PD) variability. The current study used clinical trial simulation (CTS) as a tool to investigate the effects of CYP phenotype on drug response, and to identify covariates influencing the likelihood of observing genotypic differences by integrating PK and PD information.
Dextromethorphan (DEX) was used as a model drug in CTS of antitussive response. Information on the genetics of drug metabolizing enzymes, in vitro metabolism, in vivo kinetics and the dynamics of response (drug/metabolites) was integrated. The probability of detecting a statistically significant difference in response between different CYP phenotypes (extensive (EM) and poor (PM) metabolizers) was assessed using anova. Twenty sets of CTS were carried out for each phenotype, comprising different numbers of subjects (n = 6, 22, 40, 60). The antitussive potency of the metabolite of DEX, dextrorphan, is reported to be around 40% of the parent compound [2]. In addition to the latter value, the effects of three other relative potencies (0, 100 and 200%) were examined in 480 trial simulations.
Figure 1. Power (% studies showing significant difference in response between phenotypes) vs number of subjects in each study arm.
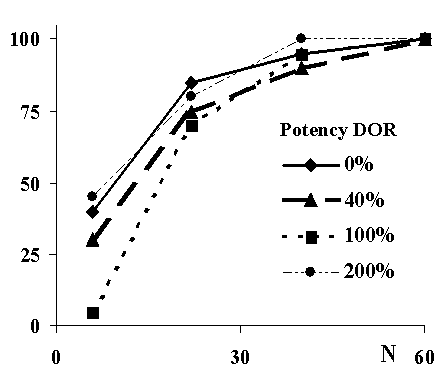
An example outcome is shown in Figure 1. At a 30 mg dose of DEX, with equal numbers of EMs and PMs in each study arm, about 30 subjects of each phenotype were required to achieve 80% power in differentiating the response between EM and PM. Greater power was evident when the metabolite was assumed to be either inactive or more potent than DEX.
Variability in PD is usually significantly greater than that in PK. Therefore, discerning any impact of genetic polymorphisms in drug metabolism on pharmacological response may be difficult even when the contribution of such metabolic routes to overall elimination is considerable, as in the current model. In addition to study size, attention should be paid to the impact of active metabolites and their activity and exposure relative to parent compound. The results from our CTS are consistent with experimental observations [3,4]. The exercise can help to rationalize inconsistent findings on the effects of genotypes on pharmacological response when study sizes are small.
1. Dickins M, Tucker GT. Int J Pharm Med 2001; 15 : 70.
2. Moghadamnia AA, et al . Br J Clin Pharmacol 2003; 56 : 57.
3. Abdul Manap R, et al . Br J Clin Pharmacol 1999; 48 : 382.
4. Capon DA, et al. Clin Pharmacol Ther 1996; 60 : 295.