
Print version
Search Pub Med

Role of COX-1, COX-2 and phospholipase A2 in prostacyclin release from aortic tissue of mature mice in vitro Prostaglandin I2 (PGI2), formed by the concerted actions of cyclo-oxygenase (COX-1 or COX-2) and prostacyclin synthase, is a potent inhibitor of platelet activation and an important cardioprotective hormone. COX-1 is present constitutively in aortic endothelium of young adult mice and is responsible for the production of PGI2 in the aortic arch (Lundberg et al. 2009, PA2 online 035P). PGI2 releasing capacity is greatly reduced in vessels from mature (12 months old) COX-1-/- mice (Lundberg et al. 2009, PA2 online 002P). However, COX-2 may be expressed in vessels with age and could therefore be associated with the cardiovascular side effects of non-steroidal anti-inflammatory drugs (NSAIDs). Here, we have extended our previous observations and compared PGI2 release from aortic tissue of mature (10-16 month old) wild type, COX-1-/- and COX-2-/- mice. We have also investigated the role of phopholipase A2 (PLA2) activation in vitro by assessing the effects of calcium ionophore, A23187, on PGI2 release from vessels of each type of mouse. Incubation of tissues with diclofenac was used to validate COX isoforms as the source of PGI2. Mice were killed by CO2 and the thoracic aorta excised, cleared of connective tissue and cut into 5 equally sized segments. Tissues were equilibrated for 60 minutes in individual wells of 96-well plates containing DMEM either with (triplicate incubations) or without diclofenac (100μM; duplicate incubations) in a humidified incubator (37°C at 5% CO2). Media was then replaced and tissues incubated for 30 minutes; samples were taken for baseline measurements, before addition of A23187 (50μM) for a further 30 minutes. Media was stored at -80°C until PGI2 was measured as its breakdown product 6-ketoPGF1α by ELISA.
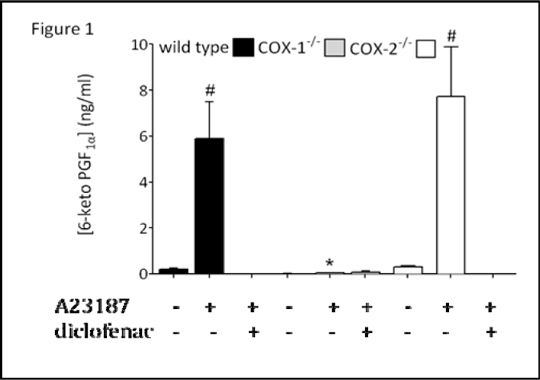
Figure 1: Release of PGI2 from aortae of wild type, COX-1-/- mice before and after addition of A23187 (50 μM) in the presence or absence of the COX inhibitor diclofenac (100 M). Data is mean ± SEM for n &%#x003D; 6 mice. Data was analysed using one-way ANOVA followed by Dunnett’s post tests; *P<0.05 for effect of genotype on A23187-stimulated release: #P<0.05 for effect of A23187, within each genotype.
Aortic tissue from wild type or COX-2-/- mice released low levels of PGI2 unless stimulated with calcium ionophore A23187 (Figure 1). A23187-induced PGI2 release was strongly inhibited by diclofenac (Figure 1). Aortae from COX-1-/- mice released negligible levels of PGI2, even in the presence of A23187. These observations support the idea that in both young and mature mice, COX-1 is responsible for the majority of vascular PGI2 production.
|
|