
Print version
Search Pub Med

How Does Pulmonary Exposure to Diesel Exhaust Particulate Prime the Heart for Increased Ischaemia/Reperfusion Injury? Episodes of increased air pollution are associated with higher cardiovascular mortality1, The adverse effects of air pollution have been attributed to particulate matter, especially ultrafine particles. Using an in vivo rat myocardial ischaemia/reperfusion (I/R) model we have previously found that infarct size, arrhythmia duration and mortality are all exacerbated after pulmonary instillation of ultrafine diesel exhaust particulate (DEP, Robertson et al., unpublished). The present study addresses the hypothesis that DEP exposure increases the sensitivity of the heart to I/R injury via stimulation of pulmonary transient receptor potential vanilloid receptor 1 receptors (TRPV1) and subsequent stimulation of the sympathetic nerve system. Male Wistar rats (250-350g, n = 3-9/group) were treated with a beta1-adrenoceptor antagonist (metoprolol, 10 mg/kg intraperitoneal) or a TRPV1 antagonist (AM9810, 30 mg/kg intratracheal) immediately before intratracheal instillation of DEP (0.5mg, Nat Inst Standards Technol (NIST)) or saline vehicle. 6 h later hearts were removed and immediately perfused with phosphate buffered saline in Langendorff mode. Coronary effluent was collected into tubes containing the spin-trip 1-hydroxy-2-carboxy-pyrrolidine (10-3M) for determination of oxidant stress generation using electron paramagnetic spin resonance spectroscopy, as previously described2. Ischaemia was induced by ligation of the left main coronary artery for 30 min followed by 2 h reperfusion. At the end of the experiment, the artery was religated and the area at risk (AAR) was determined by perfusion with Evans Blue. The extent of necrosis within the AAR was then determined by triphenyltetrazolium chloride staining. Perfusion pressure was comparable in hearts from DEP and saline instilled rats before initiation of ischaemia. Oxygen free radical generation was significantly higher in hearts from DEP-instilled (6606 ± 936 arbitrary units (AU)) compared to hearts from saline-instilled rats (2140 ± 224 AU, P<0.01), and prevented by co-administration of metoprolol (2328 ± 310 AU, P<0.01, 1 way ANOVA followed by Bonferroni post-hoc test). The AAR of the left ventricle was comparable in all treatment groups. However, the size of the necrotic zone was significantly increased in hearts from DEP- compared to saline-instilled rats (Figure 1). The influence of DEP on infarct size was prevented in hearts from rats that received metoprolol or AM9810 at the time of instillation (Figure 1).
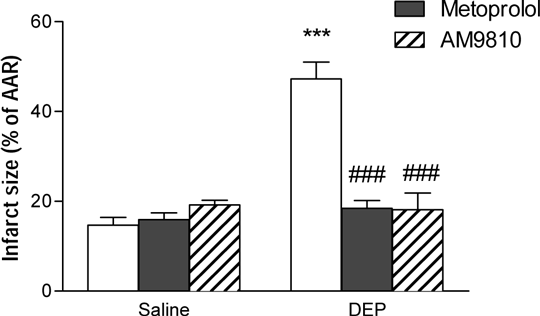
Figure 1. Myocardial infarct size (%AAR) in hearts isolated 6h after pulmonary instillation of saline or diesel exhaust particulate (DEP) alone (open bars), or with metoprolol (10 mg/kg ip, filled bars) or AM9810 (10 mg/kg intratracheal, hatched bars). Results are expressed as mean ± SEM (n = 3-9) *** P<0.001 versus saline; ###P<0.001 versus DEP; two-way ANOVA followed by Bonferroni post-hoc test. These results demonstrate that hearts are primed in vivo following pulmonary exposure to DEP, leading to increased oxidant stress and sensitivity to reperfusion injury ex vivo. Activation of a TRPV1-mediated autonomic reflex appears to be necessary for priming of the heart for subsequent injury. This mechanism may provide an additional explanation for the heightened incidence of cardiac death during periods of increased air pollution.
1 Brook RD (2008) Clin Sci; 115(6):175-187 2 Jeanes et al. (2008) J Endocrinol; 197: 493-501
|
|