Print version
Search Pub Med

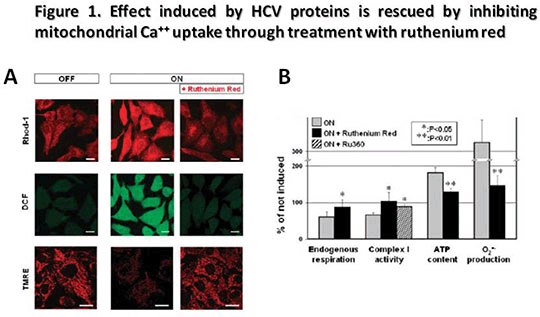
Relief of HCV-Induced Oxidative Stress by the Ca++-Uniporter Inhibitor Ruthenium Red Recent findings of co-localization of a proportion of hepatitis C virus (HCV) core protein, expressed in cultured cell lines, at physical contact sites between ER and mitochondria have put forward the possibility that Ca++ and/or oxidative stress may be involved in HCV infection. We have extended these observations by investigating the mitochondrial oxidative metabolism and intracellular calcium distribution in U2-OS human osteosarcoma-derived cell lines inducibily expressing the entire HCV open reading frame. A comparative analysis of the OXPHOS complexes activity and related ROS production along with mitochondrial morpho-functional and intracellular calcium confocal microscopy [1, 2] imaging was carried out in U2-OS cells inducibly expressing the HCV polyprotein under the control of a tetracycline-regulated gene expression system. In these cells viral protein expression is repressed in the presence of tetracycline whereas viral proteins become detectable 4-6 h and reach a steady state 24-48 h following tetracycline withdrawal [3]. Confocal microscopy analysis using specific probes revealed that cell expressing HCV proteins exhibited: i) marked mtΔΨ depolarisation (25±10 vs 50±10 A.U.); ii) increase of mitochondrial ROS production (125±15 vs 25±4 A.U.); iii) enhanced mitochondrial Ca++ load (140±12 vs 70±10 A.U.); iv) an 80% increase of ATP level as compared to controls. Moreover complex I activity as well as the endogenous respiration was significantly reduced (65±5 vs 100±6 activity % of non induced and 1,8±0,4 vs 2,9±0,6 nmoles/min/106 cells respectively) whereas complexes III and IV activities were not affected (121±24 vs 99±21 and 119±21 vs 99±23 activity % of non induced respectively). Of note, all the alterations observed were rescued to the control levels upon treatment of the infected cells with ruthenium red (Fig. 1). Our study shows a detrimental effect of HCV proteins on the cell oxidative metabolism with inhibition of respiratory chain activity and increased production of reactive oxygen species. These alteration come together with de-regulation of the calcium recycling between cytoplasm and intracellular Ca++ stores. The link appears to be causative since all the modifications observed in U2-OS cells expressing the HCV were completely reversed by the Ca++ uniporter inhibitor ruthenium red. These results, provide new insight into a possible involvement of the mitochondrial dysfunction in the pathogenesis of hepatitis C and its potential role in the development of hepatocellular carcinoma, thus suggesting alternative approaches for therapeutic strategy. Reference [1] Piccoli C, Scrima R et al, Transformation by retroviral vectors of bone marrow-derived mesenchymal cells induces mitochondria-dependent cAMP-sensitive reactive oxygen species production. Stem Cells. 2008 Nov; 26(11):2843-54. [2] Papa S, Sardanelli AM et al, Mitochondrial respiratory dysfunction and mutations in mitochondrial DNA in PINK1 familial parkinsonism. J Bioenerg Biomembr. 2009 Dec;41(6):509-16. [3] Moradpour D, Heim MK et al, Cell lines that allow regulated expression of HCV proteins: principles and application, in: R.F. Schinazi, J.-P. Sommadossi, C.M. Rice (Eds.), Frontiers in Viral Hepatisis, Elsevier B.V., Amsterdam, 2003, pp. 175–186.
 |