Print version
Search Pub Med

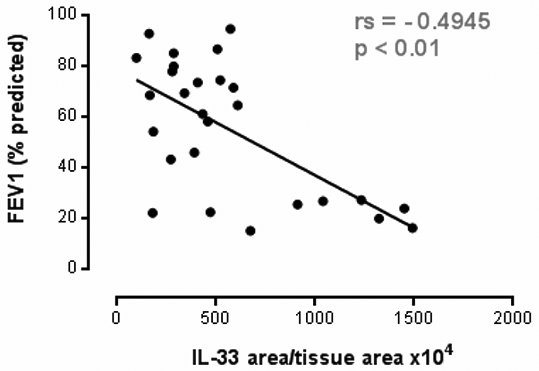
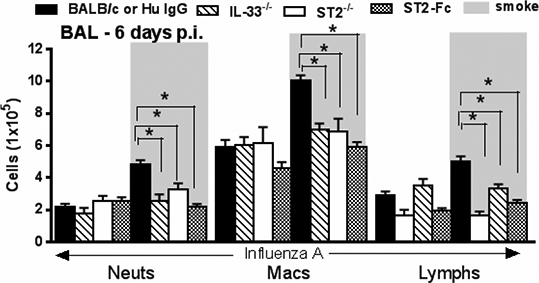
Interleukin 33: A critical mediator that orchestrates innate immune responses to virus induced exacerbations of Chronic Obstructive Pulmonary Disease Rationale: The alarmin IL-33 is an IL-1 family member primarily released in response to tissue injury and necrosis. While suggested to be involved in a plethora of inflammatory diseases, in particular Asthma, its potential role in Chronic Obstructive Pulmonary Disease (COPD) is unknown. Methods: We have used patient samples and mouse models to investigate the expression and function of IL-33 in COPD. Unless otherwise stated, data are expressed as Mean±SEM, and p values were calculated using a Mann-Whitney U test. Results: Here we postulate a novel role for the IL-33/ST2 axis in COPD by showing significantly upregulated expression, specifically within the airway epithelium of COPD patients, correlating with disease severity as measured by decline in FEV1 (Figure 1). Subsequent functional studies in mice revealed that cigarette smoke (CS) exposure significantly enhanced intracellular IL-33 levels and yielded a similar epithelial expression as to that seen in man (IL-33 positivity per total tissue area; Control:450±15 vs Smoke:551±21.1, n=8/group, p<0.05). Despite this increase, mice deficient in IL-33 or its cognate receptor, T1/ST2, were not protected from CS-induced inflammation or emphysema. However, further analyses suggested that this was due to lack of IL-33 release following smoke exposure. One of the key characteristics of COPD is debilitating pathogen-induced exacerbations and given the airway damage following viral infection, we examined the role of IL-33 in a CS-induced viral exacerbation model. Remarkably, deficiency in IL-33, its receptor or therapeutic IL-33 blockade conferred complete protection from exacerbated disease, i.e. these mice did not exhibit enhanced inflammation or exacerbated weight loss when compared to respective controls (Figure 2). IL-33 appeared to be an early mediator since we observed changes in cytokines associated with the innate immune response as early as 2 days post virus infection (significant changes in IFN-γ, IFN-α, TNF-α, IL-6, CXCL5 and IL-12p40 comparing wild type and ST2 deficient mice, n=8-16 mice/group, 1-2 experiments).
Figure 1 IL-33 expression correlates with disease severity in COPD Spearman’s rank correlation was performed, n=27. 
Figure 2 IL-33 is required for exacerbated viral inflammation due to prior smoke exposure BAL cell data are expressed as Mean±SEM, n=8-16 mice/group, 1-2 experiments, *p<0.05 
Conclusion: These data suggest that IL-33 is upregulated during COPD, released upon pathogen-induced damage and may be a key player in orchestrating acute exacerbations of COPD through its action on the innate immune system.
|