Print version
Search Pub Med

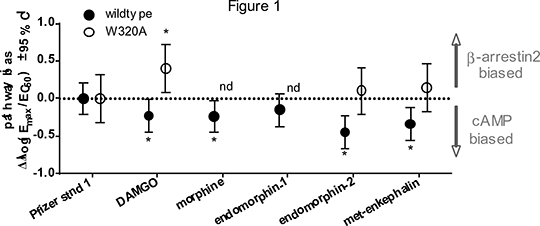
Investigating the structural determinants of µ-opioid receptor ligand bias by mutagenesis Ligand bias describes observations that some GPCR ligands can preferentially activate certain receptor pathways over others. There is growing enthusiasm for optimising ligand bias in drug discovery for anticipated pharmacotherapeutic benefit. Despite this, our ability to rationally develop novel biased ligands is hindered by an incomplete understanding of precisely how ligand binding site interactions influence differential receptor activation. The µ-opioid receptor is one example that is eagerly targeted; to develop G protein biased agonists as improved analgesics. To investigate which residues within the µ-opioid binding site play a role in generating bias, we introduced point mutations rationally informed by structural biology data. Mutant receptor pharmacology was compared to the wildtype for a panel of ligands at the G protein (cAMP assay) and β-arrestin2 (PathHunter assay) branches of signalling, in the same HEK293 cell expression system. The effect of mutation was quantified by derivation of bias factors (ΔΔlog(Emax/EC50) ± 95 % CI), using Pfizer standard-1 (2-(L-Tyrosylamino-1-[N-acetly-L-phenylalanyl)-amino]-2-methylpropane) as a reference agonist. We identified W320A as a mutant with a particularly interesting effect on opiate bias. Compared to wildtype, cAMP response maxima were reduced to 48.85 ± 13.58 %, and β-arrestin2 to 9.50 ± 0.86 %, despite similar expression levels. At the wildtype, (Figure 1; filled circles), DAMGO was moderately cAMP-biased, whilst at W320A (open circles) this switched to β-arrestin2 bias. Endomorphin-2 and met-enkephalin changed from being cAMP-biased to unbiased at the mutant. There was a selective loss of endomorphin-1 β-arrestin2 responses at W320A, despite this ligand being highly efficacious at the wildtype.
These data increase our understanding of the molecular mechanisms governing ligand bias, highlighting a role for the µ-opioid receptor binding site residue W320 in forming an active receptor conformation required for pathway-selective signalling. It is particularly intriguing that mutant receptor function changes in a ligand-specific manner. Together, this supports the hypothesis that differential receptor signalling can be regulated at the level of differential ligand/receptor interactions. The challenge now is to apply this knowledge to improve future drug discovery strategies.
|