

| 106P London, UK Pharmacology 2016 |
A role for G-protein coupled receptor 55 (GPR55) in high fat diet-induced cardioprotection
Introduction: Activation of G-protein coupled receptor 55 (GPR55) by its endogenous ligand lysophosphatidylinositol (LPI) stimulates numerous pathways, including intracellular calcium release and regulation of gene expression via ERK1/2 MAP kinase activation. Adipose tissue mRNA and protein expression of GPR55 are upregulated in obesity1 alongside elevated plasma concentrations of LPI, and mice lacking GPR55 are highly susceptible to diet-induced obesity2. We have previously shown that LPI, delivered via the coronary circulation in isolated hearts, increases infarct size (IS) following global ischaemia/reperfusion (I/R) through GPR55 activation of RhoA-ROCK3, suggesting that high circulating LPI levels may worsen the outcome from I/R. The aim of this study was to determine the effect of an obesogenic diet on I/R injury in the presence and absence of a functioning LPI/GPR55 system.
Method: Male wild type (WT; 57Bl/6J) and GPR55-/-mice were fed a standard (SD) or high fat (HFD; 40% energy from fat) diet ad libitum for 13 weeks prior to anaesthesia (ketamine and xylazine; 120mg/kg and 16mg/kg i.p. respectively). The hearts were removed, perfused retrogradely in Langendorff mode and subjected to 30 min global ischaemia followed by 30 min reperfusion. IS was measured using triphenyltetrazolium chloride (TTC) and activation of the reperfusion injury salvage kinase (RISK) pathway determined by measurement of ERK and Akt phosphorylation. Data are shown as mean±SEM (n animals) and analysis performed using two-way ANOVA followed by Bonferroni post-hoc test.
Results: GPR55-/- HFD mice exhibited significantly increased weight gain compared to mice fed a ND, while WT HFD mice did not develop an obese phenotype (Figure 1). WT HFD, but not GPR55-/-HFD, mice had significantly smaller myocardial infarcts compared to SD fed controls (Figure 2). HFD produced a modest, but not significant, decrease in cardiac ERK phosphorylation in both strains, with no effect on Akt phosphorylation.
Conclusions: Consumption of a HFD in the absence of weight gain (WT mice) decreases myocardial I/R injury through a mechanism independent of activation of the RISK pathway. The absence of a cardioprotective effect of HFD in GPR55-/- mice may be related either to the obese state of the animals, or to the loss of a functional GPR55/LPI system, implicating a protective role for endogenous cardiac LPI in the setting of acute myocardial infarction.
References:
1. Moreno-Navarette JM et al (2012). Diabetes 61: 281-291.
2. Meadows A et al (2016). Int J Obes (Lond) 40: 417-424
3. Robertson-Gray OJ et al (2014). http://www.pA2online.org/abstracts/Vol12Issue3abst160P.pdf
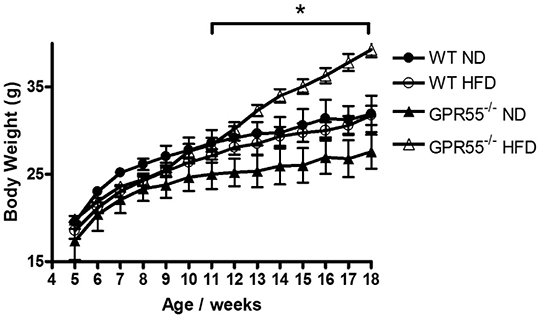 Figure 1: Change in body weights of mice over the dietary period. *p<0.05 GPR55-/- HFD vs GPR55-/- SD. n=10 per group
Figure 1: Change in body weights of mice over the dietary period. *p<0.05 GPR55-/- HFD vs GPR55-/- SD. n=10 per group
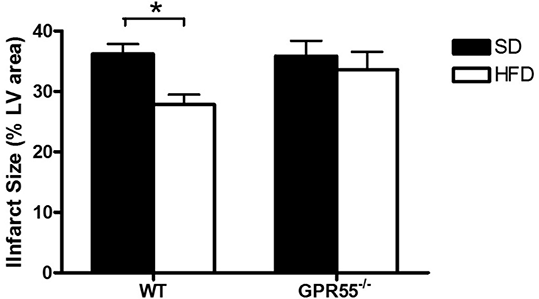
Figure 2: Effect of HFD on infarct size in hearts frow WT and GPR55-/- mice subjected to global I/R. *p<0.05 WT HFD vs WT SD. n=10 per group