| 010P London, UK 8th European Workshop on Cannabinoid Research |
Characterisation of CB1 cannabinoid receptors in molecular docking studies
Introduction: Molecular Docking is a widely used powerful tool in structure-based drug design and small molecule discovery. In the docking of proteins with highly flexible loops and overlapping binding regions such as cannabinoid receptors, the effective sampling of the conformational space is crucial. This study aimed at increasing the understanding of the efficiency of molecular docking in predicting binding sites and distinguishing ligands of the CB1 cannabinoid receptor in comparison to those observed in crystal structures.1,2
Method: Tools from the Schrodinger software suite (release version 2017-1) were used in the study. A refined model of CB1 using its crystal structure (PDB ID: 5U09)1 as template was modelled in Prime. The ICL3 loop (absent in the crystal structure) was modelled into it; N-terminus residues from 1-99 and C-terminus residues from 413-472 were omitted. The protein file was prepared using the Protein Preparation Wizard, and then energy minimised. Agonists and antagonists with established Ki values were either retrieved from PubChem database or drawn using Maestro, and prepared using the LigPrep tool. Molecular dockings were performed using Glide with extra precision (for antagonists) and Induced Fit Docking (IFD) (for agonists). GlideScore (an approximation of binding affinity) generated for each conformation in Glide and IFD and data from literature were used as criteria to predict the binding site for a ligand. They were then plotted against the corresponding experimental pKi values, and the Spearman's rank correlation coefficient (ρ) and coefficient of determination (R2) were calculated.
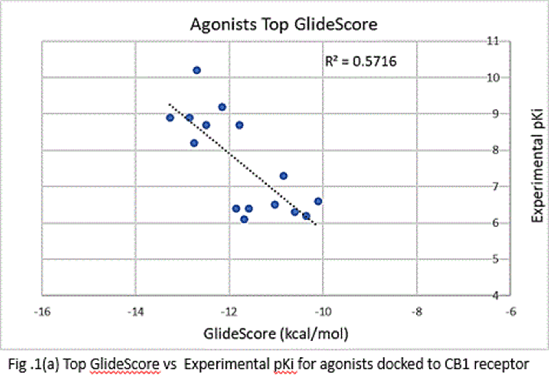
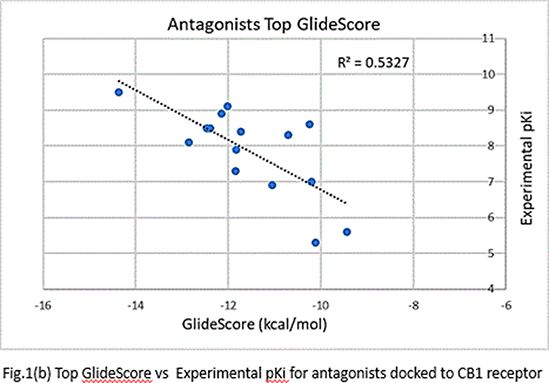
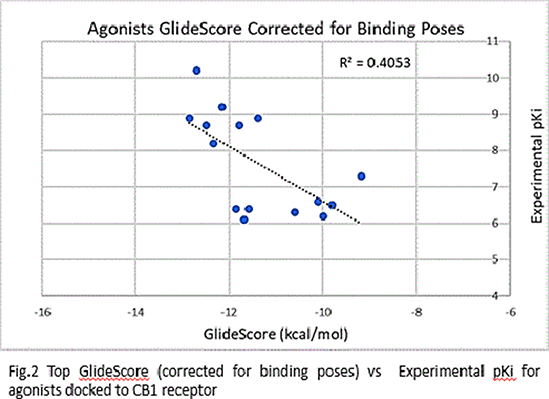
Results: The ρ and R2 values for the top GlideScores vs. experimental pKi values were -0.7560 and 0.5716 respectively for agonists (Fig.1(a)) and -0.729 and 0.5327, respectively, for antagonists (Fig.1(b)). However, upon correcting the GlideScores for realistic binding sites for agonists, the ρ and R2 changed to -0.6366 and 0.4053, respectively, for agonists(Fig.2). Correction for antagonists was not required.Although the binding positions of most ligands were like those obtained in docking of CB1 crystal structures, the orientation of aminoalkylindoles and ∆8-THC analogues were markedly different.
Conclusions: The data obtained indicate that the docking protocols chosen distinguish well between agonists and antagonists bound to CB1 receptors. The correlation between the top scores and binding conformation is better in the case of antagonists than agonists.
1.Shao et al. (2016) Nature PMID:27851727
2.Hua et al. (2016) Cell PMID: 27768894