

| 035P London, UK Pharmacology 2017 |
Modelling biased agonism dynamics at G protein-coupled receptors
Introduction: Theoretical models of G protein-coupled receptor (GPCR) concentration-response relationships often assume an agonist producing a single functional response via a single active state of the receptor. These models have largely been analysed assuming steady-state conditions. There is now much experimental evidence to suggest that many GPCRs can exist in multiple receptor conformations and elicit numerous functional responses, with ligands having potential to activate different signalling pathways to varying extents - a concept known as biased agonism. Moreover, recent experimental results indicate a possibility for time-dependent bias, whereby an agonist\\'s bias with respect to different pathways may vary dynamically1. Efforts towards understanding the implications of temporal bias by characterising and quantifying ligand effects on multiple pathways will benefit from extending current equilibrium binding models to include G protein activation dynamics.
Method: We formulate a new mathematical model of time-dependent biased agonism, based on ordinary differential equations for multiple cubic ternary complex activation models with G protein cycle dynamics. The model is generally applicable to systems with NG G proteins and N* active receptor states. Numerical simulations with NG=N*=2 are performed to generate time courses of active Gα subunits to indicate a response downstream of the receptor. Effects of efficacy and cooperativity parameters on Gα dynamics are investigated, and related to bias factors calculated using an existing definition2.
Results: Simulations give new insights into the effects of system parameters on bias dynamics, revealing new phenomena including:
• dynamic inter-conversion of ligand effect, whereby a Gα level may dynamically change from above-basal to sub-basal;
• non-monotonic concentration-response curves for single readouts and functionally antagonistic responses based on competing downstream pathways;
• dynamic inter-conversion of bias direction, where the pathway to which the ligand is apparently biased may change dynamically.
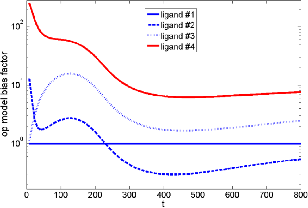
Figure 1: Simulated bias factors (with respect to two Gα signals, ligand #1 as reference ligand) for four ligands, varying dynamically. Further, our model recapitulates experimental data for two competing G protein pathways (Gi and Gs) that become activated upon stimulation of the adenosine A1 receptor with adenosine derivative compounds.
Conclusions: Our model supports the recent suggestion that kinetic context of biased signalling should be considered in quantifying and understanding biased agonism. We propose this model as a theoretical framework for further study of the potential benefits of developing biased agonists as therapeutics.
References:
1. Herenbrink CK et al. (2016). Nature Communications 7.
2. Kenakin T et al. (2012). ACS Chemical Neuroscience 3(3):193-203.