
Print version
Search Pub Med

| 072P London, UK Pharmacology 2017 |
Dysfunctional NMDA receptors in neurodevelopmental disorders
Introduction: Genome-wide sequencing has identified mutations in N-methyl-D-aspartate receptor (NMDAR) subunits as the only disruptions in patients exhibiting a variety of neurodevelopmental disorders1. Here, we focus on four (C461F, P553L, N615I, V618G) de novo missense mutations in the GluN2B subunit, which exhibits high expression levels during early stages of development. We predicted that these mutations would have pathophysiological implications and therefore examined their effects on NMDARs.
Methods: GluN2B mutants were generated using site-directed mutagenesis and expressed in transfected HEK293 cells with GluN1 and eGFP. Cultured dissociated hippocampal neurons were prepared from E18 Sprague-Dawley rats and transfected with GluN2B cDNAs. Whole-cell electrophysiology was performed, data analysis used a new kinetic model for the NMDAR. Data are stated as mean±SEM, and analysed using one-way ANOVA with Dunnett’s post-hoc test. A homology model of the human GluN1-GluN2B receptor was generated using NMDAR crystal structures as templates2,3. Mg2+ and memantine binding sites were defined by docking studies.
Results: The human NMDAR homology model located the mutant residues. Electrophysiology revealed loss/gain-of-function phenotypes. Notably, the ion channel mutants, N615I and V618G, ablated Mg2+ voltage-dependent block, with distinct effects on divalent cation permeation (Table 1). In the ligand binding domain, C461F, reduced glutamate potency, and P553L markedly increased the rate of NMDAR desensitisation compared to wild-type receptors (Table 1). In neurons, these mutations minimally affected the synaptic current amplitude although the decays were often accelerated (Table 1). We attempted to reverse the gain-of-function phenotype with the channel blocker memantine. The N615I and V618G mutant NMDARs exhibited some voltage dependence with significantly altered sensitivity to memantine (IC50 N615I 1.5±0.3 μM; V618G 52.2±3.7 μM, WT 7.3±1.9 μM, n=5-7, P<0.05). These data were integrated with a new structural model showing Mg2+ and memantine binding in the NMDAR ion channel.
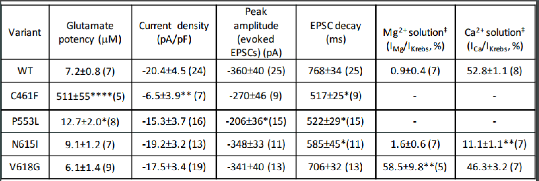
Table 1 Functional effects of GluN2B subunit mutations. *p<0.05; **p<0.01; ****p<0.0001 vs control from (n) experiments. ‡External solution where Mg2+ and Ca2+ are the only current (I)-carrying ions.
Conclusions: The functional effects of the missense mutations revealed gain/loss-of-function that affected NMDAR ligand binding and ion channel properties. These profiles may underpin epileptic phenotypes of patients that carry the mutations. Moreover, the study indicates that memantine could be a promising treatment for selected gain-of-function mutations in NMDA receptors.
References:
1. Burnashev N and Szepetowski P (2014) Curr Opin Pharmacol 20: 73-82
2. Lee C-H. et al. (2014) Nature 511: 191-197.
3. Karakas E and Furukawa H (2014) Science 344: 992-997.