Print version
Search Pub Med

| 118P London, UK Pharmacology 2017 |
Agonist-dependent inhibition of arrestin-3 recruitment to μ-opioid receptor by novel GRK inhibitors
Introduction: G-protein coupled receptor kinases (GRKs) are key regulators of GPCR signalling (1), and the search continues for potent and selective GRK inhibitors. Compound101, a GRK inhibitor, was developed by Takeda (1, 2), and it exhibits some selectivity for GRKs 2 and 3 (3). Here we have synthesised some novel compound101 analogues, with the aim of determining their abilities to inhibit agonist-induced arrestin recruitment to the μ opioid receptor (MOPr; as a proxy of GRK-induced phosphorylation of the receptor). In particular we investigated the importance of the pyridinyl-triazole moiety and the role of hydrogen bond donor and acceptor groups in their activity.
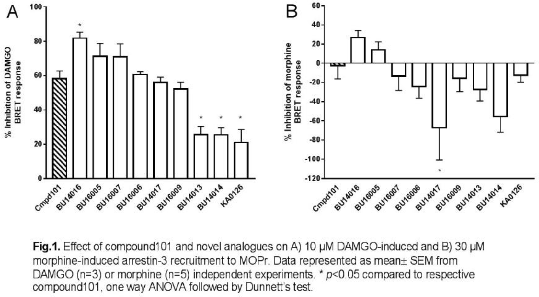
Methods: HEK293 cells were transfected with BRET donor (MOPr-RlucII) and acceptor (arrestin3-GFP) constructs. On the experimental day, cells were detached from the dish and re-seeded onto white, 96-well plates. Cells were preincubated with compound101 (100 μM) or analogues (100 μM) for 30min at 37°C. DAMGO (10 μM) or morphine (30 μM) was then added for 10 min before BRET measurement. Coelenterazine 400a (final concentration of 5 μM) was prepared 3∼6 min before BRET measurements. All the BRET measurements were performed using a FLUOstar Omega (BMG LABTECH,Germany) microplate reader with a filter set of 410/80 nm (donor) and 515/30 nm (acceptor). The BRET ratio was determined by dividing the intensity of acceptor luminescence emission at 515 nm by the intensity of donor luminescence emission at 410 nm.
Results: In DAMGO-stimulated cells, only compound BU14016 at 100 μM showed a significantly greater inhibition of arrestin-3 recruitment than compound101 (Fig.1A), whilst compounds BU14013, BU14014 and KA0126 showed significantly less inhibition. In contrast, in morphine treated cells, compound101 did not inhibit arrestin-3 recruitment, whilst only compounds BU16005 and BU14016 had any inhibitory capacity (Fig.1B). Some compounds even appeared to express stimulatory activity in the presence of morphine.
Conclusion: Most of the novel GRK inhibitors reduced DAMGO-induced arrestin-3 recruitment to MOPr. Interestingly, they were much less effective at reducing morphine-induced arrestin-3 recruitment. One possibility is that different GRKs might mediate phosphorylation of MOPr, depending upon the agonist used.
References
1. Guccione M et al. (2016). J Med Chem 59: 9277-9294.
2. Homan KT and Tesmer JJ (2015). ACS Chem Biol 10: 246-56.
3. Thal DM et al. (2011). Mol Pharmacol 80: 294-303.