Print version
Search Pub Med

| 043P Nottingham, UK 7th Focused Meeting on Cell Signalling |
Novel GRK inhibitors reduce arrestin recruitment to the mu opioid receptor
Introduction: GRKs are key regulators of GPCR signalling (1), and the search continues for potent and selective GRK inhibitors, for example for the treatment of congestive heart failure. Compound101, a GRK inhibitor, was developed by Takeda (2), and it exhibits some selectivity for GRKs 2 and 3 (3). Here we have synthesised some novel compound101 analogues, with the aim of determining their abilities to inhibit agonist-induced arrestin recruitment to the μ opioid receptor (MOPr; as a proxy of GRK-induced phosphorylation of the receptor). In particular we investigated the importance of the pyridinyl-triazole moiety and the role of hydrogen bond donor and acceptor groups in their activity.
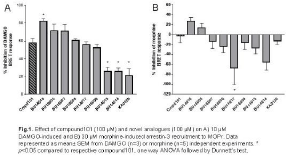
Methods: HEK293 cells were transfected with the BRET donor (MOPr-RlucII or DOPr-RlucII) and acceptor (arrestin3-GFP) constructs. On the experimental day, cells were detached from the dish and re-seeded onto white, 96-well plates. Cells were incubated with compound101 (up to 100 μM) or analogues (up to 100 μM) for 30min at 37°C. For MOPr, DAMGO (10 μM) or morphine (30 μM) was then added for 10 min before BRET measurement; for DOPr, SNC80 (10 μM) was added. Coelenterazine 400a (5 μM) was prepared 3~6 min before BRET measurements. All the BRET measurements were performed using a FLUOstar Omega (BMG LABTECH,Germany) microplate reader with a filter set of 410/80 nm (donor) and 515/30 nm (acceptor). The BRET ratio was determined by dividing the intensity of acceptor luminescence emission at 515 nm by the intensity of donor luminescence emission at 410 nm. Statistical analysis was performed using one way ANOVA followed by Dunnett’s test.
Results: In DAMGO-stimulated cells, only compound BU14016 at 100 μM showed a significantly greater inhibition of arrestin-3 recruitment than compound101 (Fig.1A), whilst compounds BU14013, BU14014 and KA0126 showed significantly less inhibition. In contrast, in morphine treated cells, compound101 did not inhibit arrestin-3 recruitment, whilst only compounds BU16005 and BU14016 had any inhibitory capacity (Fig.1B). Some compounds (BU14017) even appeared to express stimulatory activity in the presence of morphine.
Conclusion: Most of the novel GRK inhibitors reduced DAMGO-induced arrestin-3 recruitment to MOPr. Interestingly, they were much less effective at reducing morphine-induced arrestin-3 recruitment. One possibility is that different GRKs might mediate phosphorylation of MOPr, depending upon the agonist used.
References:
1. Hilger D et al. (2018). Nat Struct Mol Biol 25:4-12.
2. Homan KT and Tesmer JJ (2015). ACS Chem Biol 10: 246-56.
3. Thal DM et al. (2011). Mol Pharmacol 80: 294-303.