The
slopes of concentration-effect (dose-response) curves or
Clark, Gaddum, Schild, and pA2
Bioassay
The development of pharmacology
between the two world wars owes much to the need to measure the activity
of drugs, which had not been obtained chemically pure, in terms of international
standards. Originally this included substances such as digitalis, thyroxine
and insulin and even in 1947 extracts of curare alkaloids ('Intocostrin')
were standardised by bioassay. An important reference book was J.H.Burn's
'Biological Standardisation' (Oxford Medical Publications, 1937) and
students in his laboratory rapidly learnt that the slopes of dose-response
curves were all different - and that you needed a preparation with a
steep dose-response curve* to make a sensitive bioassay.
Differences between Pharmacology and Biochemistry: Agonists
The fact that dose-response curves varied so much made pharmacology
different from biochemistry. According to Michaelis-Menten kinetics
the substrate, S, and enzyme, E, combined one-to-one forming
the complex, ES, and the velocity of the reaction, V,
was directly proportional to ES, rising from zero to a maximum,
Vmax. The concentration of substrate producing a half-maximal
rate, Km (the Michaelis constant), half-saturates
the enzyme and is the dissociation constant for the process E + S<->ES.
The relation can be written
V = VmaxS/(S + Km) (1)
and when V is plotted against log[S] the curve is sigmoid
with central part approximately linear and with a fixed slope: to go
from V = 25% to V = 75% of Vmax, S must
be increased 9-fold. Pharmacologists working with tissues found that
a similar change from 25% to 75% of the maximum response was often produced
by much smaller increases: sometimes doubling the concentration was
sufficient. The activity of a substrate for an enzyme was determined
by Km and Vmax. In contrast, the concentration
of a drug which produced a half-maximum response from a tissue, EC50,
was not constant: with powerful agonists a maximum response was produced
from the activation of only a small proportion of receptors.
* This term was used in
the older work: it has since been replaced by 'concentration-effect'
curve.
Antagonists:
(i) effects expressed as a fraction of control responses
For biochemists studying the effects of an inhibitor, I, competing
with a substrate, S, for an enzyme, the concentration of enzyme-substrate
complex, ES, is:
ES = (S/ Km) /[1 + (S/Km) + (I/Ki)]
(2)
where Ki is the dissociation constant for the process E + I
<->EI. The reaction rate is directly proportional to ES,
so if this is measured for different concentrations of S and I
and Km is known, it is possible to estimate the inhibitor
constant, Ki. The same relation applies to the drugs
A (agonist) and B (antagonist) acting on tissue receptors
(Gaddum, 1937), but with tissues the response obtained is not directly
proportional to receptor occupancy (the receptor equivalent of ES).
So although pharmacologists can obtain concentration- effect curves for
an antagonist by measuring its effect as the inhibition of control responses
from the agonist on the tissue, the position and shape of these depend
upon the concentration of agonist used and the shape of its concentration
- effect curve. A 50% reduction in the response to a concentration of
agonist where the curve is steep represents a much smaller antagonism
than for an agonist where it is flat.
(ii) effects expressed in terms of concentrations producing the same
response
The idea of measuring the effects of an antagonist in terms of how much
you needed to increase the agonist to restore the response to its uninhibited
size appeared in a paper by Clark and Raventos (1937: Table 1).
TABLE 1
Antagonism of acetylcholine (Ach) and Me4N+
Preparation
| |
Rat intestine
|
Frog auricle
|
Frog rectus
|
| antagonist |
Ach
|
Me4N+
|
Ach
|
Me4N+
|
Ach
|
Me4N+
|
| Atropine |
-8.1
|
-7.9
|
-8.3
|
-7.7
|
-4.2
|
-3.8
|
| OctN+Me3 |
-5.5
|
-5.0
|
-5.0
|
-4.6
|
-4.1
|
-4.0
|
| Bu4N+ |
-3.6
|
-3.7
|
-
|
-
|
-3.2
|
-3.0
|
| 'curarine' |
inactive
|
inactive
|
-5.1
|
-4.5
|
-6.8
|
-6.5
|
| Figures indicate the logarithm of the molecular concentration
of antagonist which necessitated a 10-fold increase in the concentration
of Ach or Me4N+ in order to obtain the same
effect: 'curarine' was a crude extract of alkaloids. For competition
the numbers should depend on the tissue and the antagonist and not
on the agonist. |
Clark died in 1941 but after the war Schild (1947), using home-made automated
equipment, showed how antagonist activity could be measured in this way.
The responses to the agonist in the presence and in the absence of antagonist
are the same (it is a 'null' method) so ES or agonist-receptor
occupancy in equation 2 will be the same and the relation between
this and the response will not matter. He coined the phrase 'dose-ratio'*
to express the effect of a particular concentration of an antagonist.
For a dose-ratio X, the response produced by [A] of agonist (dissociation
constant KA) is also produced by X[A] in the
presence of [B] of antagonist (dissociation constant KB
), so
(A/ KA) /[1 + (A/KA)] = X(A/ KA)
/[1 + X(A/ KA) + (B/ KB)]
or X = 1 + (B/KB) (3)
This
is often called the Gaddum-Schild equation and shows that the concentration
of antagonist which necessitates doubling the concentration of agonist
(X = 2) to obtain the same size of response should be KB in
the process B + R <->BR. The symbol pA2
is the logarithm of the reciprocal of this concentration, log.[1/ KB],
a positive number which should be independent of the agonist, providing
it acts at the receptor R. It is exactly the same as biochemical
pharmacologists measure in binding experiments with receptor material
and an inhibitor competing with a radio-labelled ligand. In these, too,
the dissociation constant for the inhibitor should be the same for all
ligands and that particular receptor.
* I have kept this term although 'concentration-ratio' is in present
use.
Relation between dose-ratio and antagonist concentration (Schild
plots)
In practice it is often easier
to measure higher effects than dose-ratios of 2: it is also more accurate
because errors affect [dose-ratio - 1]. Clark and Raventos worked with
dose-ratios of 10. If a range of concentrations of antagonist is tested,
the graph of log.[DR-1] against log.[B] - a 'Schild' plot - should be
a straight line with a slope of unity if the agonist and antagonist act
competitively (Arunlakshana and Schild, 1949). As in biochemistry, antagonism
in pharmacology is not restricted to competition. Some compounds bind
covalently to a receptor. When used against an agonist with very high
efficacy - i.e. which produce a maximum response even when only a small
proportion of receptors is activated - these 'irreversible' antagonists
give linear Schild plots over a range. In pharmacology it may also be
possible to obtain the opposite effect from a tissue by a separate mechanism,
for example to antagonise contraction by causing relaxation. This has
been called 'physiological' antagonism*. A check that agonist and antagonist
are acting competitively can be made by testing them along with another
known competitive antagonist.
* The term 'inverse agonist' is used to describe the situation where
the two drugs act on the same receptor and the receptor mechanism can
operate in either direction.
Uses
The measurement of the activity of an antagonist as a log.equilibrium
constant, whether it is called pA2 or log.[1/ KB]
has obvious advantages in the testing of antagonists. It is directly proportional
to the free energy of binding (- G = RT lnKB) and errors in
its measurement appear to be normally distributed. As well as for comparing
different antagonists, it can be used for comparing the types of receptors
in different tissues or for checking that the binding characteristics
of receptor material obtained from membranes are the same as for receptors
in functional tissues. G = RT lnKB) and errors in
its measurement appear to be normally distributed. As well as for comparing
different antagonists, it can be used for comparing the types of receptors
in different tissues or for checking that the binding characteristics
of receptor material obtained from membranes are the same as for receptors
in functional tissues.
Measuring the steepness of concentration-effect curves
The relation between effect, E, and concentration, A, can
be represented:
E = M [A]P/([A]P + [A50]P)
(4)
where M is the maximum, [A50] produces half this,
and the exponent, P, is a convenient measure of the steepness.
Parker & Waud (1971) used this as an empirical model for concentration
- effect curves but it has the same form as the equation used by Hill
(1913) to describe the binding of oxygen to haemoglobin: P is the
Hill coefficient. The steepness can be estimated from the ratio of the
concentrations producing 75%, [A75], and 25%, [A25],
of the maximum: [A75]/ [A25] = 91/P
but Parker & Waud showed that with iterative calculations using a
computer it was possible to fit values of E by least-squares directly
to values of A and obtain estimates of M, P and [A50].
For one-to-one binding P=1 and the relation becomes the same as
equation (1). The exponent might indicate the molecularity of the process.
For P=2 the process might be: 2A+R<->AR: for P=0.5 it might
be: A+2R<->RAR, but, as with the binding of oxygen to haemoglobin,
the situation is unlikely to be as simple as this. Nevertheless values
of P>1 indicate that the signal generated by an agonist combining
with a receptor is amplified, and values of P<1 that it is reduced.
What you find experimentally
Values of P were calculated from figures appearing in the British
Journal of Pharmacology (vols 120-123: Barlow, 1999). The selection was
arbitrary but the distribution appeared to be log.normal: the coefficient
of skewness was 5.0 for values of P (kurtosis 52) compared with -0.04
(kurtosis 4.5) for values of log.P, and skewness = 0 (kurtosis 3) for
a normal curve. The results are summarized in Table 2.
TABLE 2
Values of the exponent, P
| |
range
|
sd
|
mean
|
log. mean
|
median
|
n(*)
|
| 1 |
Binding experiments |
| |
0.18-1.30
|
0.26
|
0.74
|
0.68
|
0.75
|
51 (8)
|
| 2 |
Measurements of electrical
events, e.g.currents |
| |
0.29-2.33
|
0.54
|
1.21
|
1.09
|
1.04
|
41 (4)
|
| 3 |
Measurements of second
messengers |
| |
0.25-3.67
|
0.65
|
1.00
|
0.86
|
0.84
|
64 (9)
|
| 4 |
Positive effects on tissues,
e.g. contraction |
| |
0.33-8.02
|
0.88
|
1.12
|
0.99
|
0.97
|
81 (3)
|
| 5 |
Negative effects on tissues,
e.g. relaxation or inhibition |
| |
0.20-2.12
|
0.43
|
1.05
|
0.96
|
0.96
|
118 (6)
|
|
* number with P<0.5
|
For binding experiments (1) P should be 1and there are many
examples in the literature (outside the survey) where this is so. In this
sample there was no value greater than 1.3 but a long 'tail' of values
less than 1, with 8 out of 51 (16%) having P<0.5. Some of the
flat curves involved two binding sites and with others it appeared that
the baseline was greater than zero.
In groups 2 and 3 the events measured should be closer to the binding
step than those in groups 4 and 5. From the range it appears that amplification
of the signal can occur and the biggest effects are with positive effects
on tissues (4). The sample, however, has not included some curves which
were known to be very steep long before computer methods were available
for estimating P. The inhibition of contractions of the phrenic
nerve-rat diaphragm preparation by (+)tubocurarine chloride is an example.
Re-examination of some old results gave P=3.74, greater than the
biggest value for negative responses (group 5 in Table 1) but it is known
that the compound competitively antagonizes neurally released acetylcholine.
Why is the curve so steep? The shapes and position of curves produced
by competitive antagonists depend upon the size of the agonist effect
used in the experiments - this can be called the 'degree of agonist stimulation'
and expressed as A/A50. When the antagonist opposes
a small effect the value of P is close to 1. When a large effect
is used the curve is shifted towards higher concentrations and P
is that for the agonist. In the experiments with (+)tubocurarine chloride
it appears that neural stimulation produces a very high agonist effect
indeed with A/A50 approximately = 12 (Barlow, 1995).
If antagonist concentration - inhibition curves are obtained where each
concentration of antagonist was tested against both a low (A/A50<
1) and a high (A/A50>1) degree of agonist stimulation
it is possible to calculate antagonist dissociation constants, KB
(Barlow et al. 1997). This could be a useful alternative to measuring
dose-ratios when the antagonist has limited solubility.
Operational windows
(i) agonists
The steepness of concentration-effect curves may depend upon the way the
effect is measured. In order to study this parallel experiments were made
with contractions recorded isotonically (proportional to the change in
length) and isometrically (proportional to the change in tension), using
tissues from the same animal and with the same load or initial tension
(Barlow et al, 2001a). For contraction of the rat uterus in oestrus by
carbachol* the mean values of P (n 10) were 7.7 (se 1.9, isotonic)
and 1.9 (se 0.2, isometric). In experiments with guinea-pig ileum mean
values (n 13) were 2.49 (se 0.22, isotonic) and 1.73 (se 0.19, isometric)
for carbachol: for histamine (n 7) they were 1.74 (se 0.20, isotonic)
and 1.29 (se 0.14, isometric). In all these experiments the curves with
isotonic recording were also displaced towards lower concentrations. Examples
are shown in Figure 1. An explanation for the higher values of P
with isotonic recording is that the 'operational window' - between what
is recorded as zero and as maximum - is narrower. It is possible to record
increases in tension with the tissue held at a fixed length when the muscle
cannot shorten further because of its bulk.
(ii) antagonists
Many of the curves in group 5 are for substances, e.g. relaxants or dilators,
physiological antagonists which activate separate receptors producing
the opposite effects to constrictors. These are particularly likely to
be affected by the operational window of the recording system. With isometric
recording it will be possible to measure decreases in tension beyond the
point where the tissue is fully extended under the load. When relaxation
curves were obtained for (-)isoprenaline opposing the effects of carbachol
or histamine on guinea-pig ileum, values of IC50 were lower
with isotonic than with isometric recording in parallel experiments (Barlow
et al, 2001b). The curves were steeper and there were wider shifts
in antagonist concentration before the point was reached where increasing
the concentration of agonist produced a contraction which could not be
overcome by the antagonist. To standardise methods for comparing drugs
and avoid the complications arising from the shape of the agonist curve,
the effects of antagonists can be compared against agonist concentrations
which on their own produce a half-maximum effect.
(iii) windows and signal amplification
The idea of operational windows can be extended from recording systems
to cellular events close to the response. The highest value in Table 1,
P=8 (Shiraishi et al1998), is for the relation between tension
in the middle cerebral artery of the rabbit and the internal concentration
of calcium ions. There is a narrow window between concentrations which
produce no effect on tension and those which produce the maximum: narrow
windows produce steep concentration - effect curves. If the signal curve
generated by the previous step is represented by equation 4 and only the
points lying within the window are fitted to this expression, it is possible
to see that the curve is not symmetrical. If there is a threshold, the
lower half will be steeper than the overall fit and the upper half flatter.
The high values of P indicate selection rather than amplification
of the signal. Lew and Flanders (1999) call this 'threshold inertia' and
it should be particularly important for the effects of physiological antagonists
which probably ultimately act by raising or lowering the internal concentration
of calcium ions.
* Widely used for the assay
of adrenaline (which causes relaxation) before spectrophotofluorimetric
methods were invented.
Future Developments
This article was inspired by the choice of 'pA2' as
a title replacing 'BPS Bulletin' and the need for Graeme Henderson to
explain pA2 for the benefit of 'members of the society
who have entered pharmacology from another discipline'. It is such people,
anatomists, biochemists, biophysicists, chemists, pathologists, pharmacists,
physiologists, statisticians and, above all, medically qualified people
who in the past have made pharmacology what it is. Gaddum referred to
a pharmacologist as a Jack (Jill)-of-all-trades and it is important to
retain this outward-looking approach. Perhaps what distinguishes pharmacologists
is that they do not limit the compounds they test to those which occur
naturally and that they are concerned with comparing the activities of
different compounds, so far as is possible. The wide variation in the
slopes of concentration - effect curves is a particular feature of pharmacology
which makes this difficult and the title pA2 is a fitting
tribute to past work on the problem of measuring the activity of competitive
antagonists. It is to be hoped, however, that this will be seen in context:
much more remains for the future.
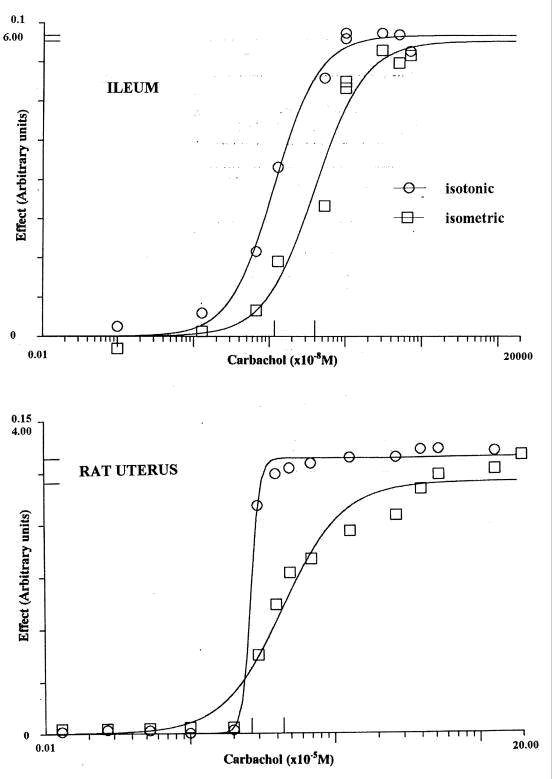
Figure
1. Concentration-effect curves for carbachol with samples of adjacent
tissue from guinea-pig ileum and rat uterus in oestrus recorded isotonically
and isometrically. The scales have been chosen so that the graphs are
of roughly comparable height with the maxima indicated on the ordinate:
values of EC50 are marked on the abscissa.
References
Arunlakshana, O. & Schild, H.O, (1949) Some quantitative uses of drug
antagonists. Br. J. Pharmacol., 14, 48-58.
Barlow, R.B. (1995) The use of an antagonist for estimating the degree
of agonist stimulation during physiological release. Trends Pharmacol.
Sci., 16, 262-264.
Barlow, R.B. (1999) A survey of the slopes of concentration- response
curves. Br. J. Pharmacol., 128, 301P.
Barlow, R.B., Susan M. Bond, Elizabeth Bream, Lisa Macfarlane, & McQueen,
D.S. (1997) Antagonist inhibition curves and the measurement of dissociation
constants. Br. J. Pharmacol., 120, 13-18.
Barlow, R.B., Susan M. Bond, Claire Grant, D.S.McQueen & Zeenat Yaqoob
(2001 a) A comparison of effects measured with isotonic and isometric
recording: I Concentration- effect curves for agonists. Br. J. Pharmacol.,
133, 1081-1086.
Barlow, R.B., Susan M. Bond, Claire Grant, D.S.McQueen & Zeenat Yaqoob
(2001 b) A comparison of effects measured with isotonic and isometric
recording: II Concentration- effect curves for physiological antagonists.
Br. J. Pharmacol., 133, 1087-1096.
Clark, A.J. and Raventos, J. (1937) The antagonism of acetylcholine and
of quaternary ammonium salts Quart. J. Exp. Physiol., 26, 375-392.
Gaddum, J.H. (1937) The quantitative effects of antagonistic drugs. J.
Physiol., 89, 7P-9P.
Hill, A.V. (1913) The combinations of Haemoglobin with Oxygen and Carbon
Monoxide I. Biochem. J., 7, 471-480.
Lew, M.J. & Flanders, S. (1999) Mechanisms of melatonin-induced vasoconstriction
in the rat tail artery; a paradigm of weak vasoconstriction. Br. J. Pharmacol.,
126, 1408-1418.
Parker, R.B. & Waud. D.R. (1971) Pharmacological estimation of drug-receptor
dissociation constants. Statistical evaluation. I Agonists J. Pharmacol.
Exp. Ther., 177, 1-12.
Schild, H.O. (1947) pA, a new scale for the measurement of drug antagonism.
Br. J. Pharmacol., 2, 189-206.
Shiraishi, Y, Kanmura, Y. & Itoh, T (1998) Effect of cilostazol, a
phosphodiesterase type III inhibitor, on histamine-induced increase in
[Ca2+]i and force in middle cerebral artery of the
rabbit. Br J.Pharmacol., 123, 869-878.
R. B. Barlow
|

